Introduction about Dr. Scott L. Friedman
Hello, this is Dr Scott Friedman at the Icahn school of medicine at Mount Sinai. I’m delighted to give this lecture on behalf of the PanNASH initiative. My title is HCC pathogenesis in NASH and the impact of fibrosis. This is adapted from a lecture I gave recently to the Paris NASH meeting, and I’m pleased to illustrate the highlights of that topic in this talk today. I have many commercial interactions, none of which are related directly to the content of this talk, but I list them for completeness’ sake.
Overview of HCC Pathogenesis in NASH and the Impact of Fibrosis
Here’s the overview of my lecture. I will cover three areas of HCC fibrosis in NASH:
Table of Contents
- HCC/Fibrosis in NASH – Epidemiology and Clinical Features
- HCC Mechanisms that are Unique to NAFLD or Non-Alcoholic Fatty Liver Disease and not Fibrosis-Dependant
- Livers Has the Highest Relative Risk of Cancer Due to Obesity
- Mechanisms of Increased Cancer Risk Associated with Obesity
- NASH-Dependent Immune Response in HCC
- Article: NASH Limits Anti-Tumor Surveillance in Immunotherapy-Treated HCC
- Genetic Contribution of PNPLA3 to NASH HCC
- Genetic Polymorphisms Linked To NASH-HCC
- HCC Development in Western Diet/CCI4 FAT-NASH (“Fibrosis and Tumors”) Model
- Mechanisms Linking Fibrosis directly to NASH-HCC Hepatocellular Carcinoma
- NASH-HCC+ Fibrosis – Summary
HCC/Fibrosis in NASH – Epidemiology and clinical features
NAFLD – Spectrum of Hepatic Pathology
This is the spectrum of non-alcoholic fatty liver disease; it includes patients with fat alone, known as non-alcoholic fatty liver. Up to 40% of Americans have this condition, of whom 25% progress to this state of non-alcoholic steatohepatitis. This is the disease stage at which there is great attention being focused both because of its risk of inflammation and progressive fibrosis and cirrhosis shown here, but also because of the risk of hepatocellular carcinoma.
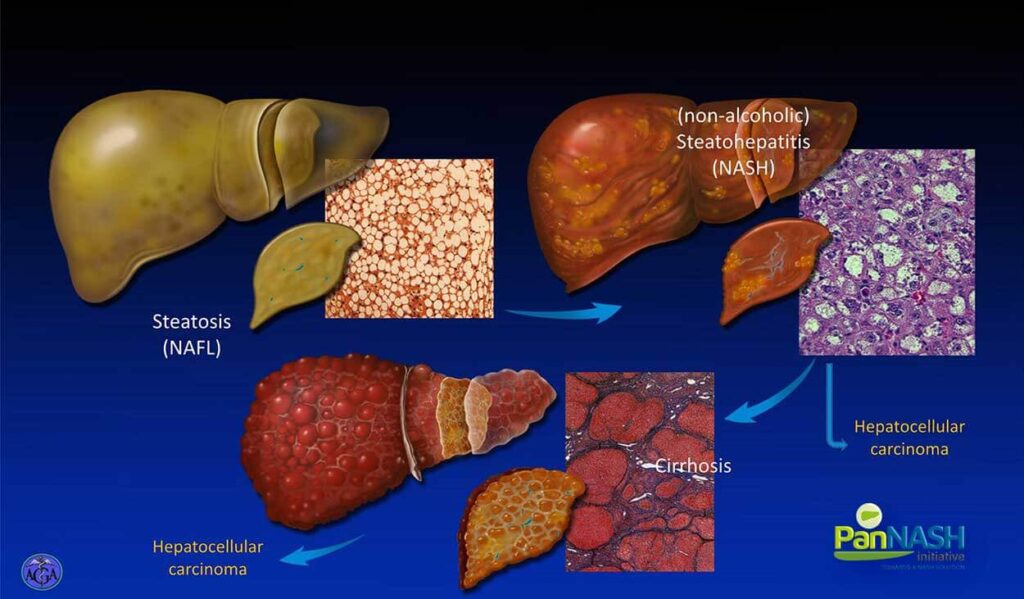
I will all also emphasize very shortly the unique feature of HCC and NASH, which suggests that up to a third of patients will develop hepatocellular carcinoma while they still have NASH but before they have developed cirrhosis, so we need to be vigilant about the risk of HCC earlier in the stages of disease for underlying NASH compared to viral hepatitis.
Nash is the Fastest Growing Cause of HCC Among Livers Diseases
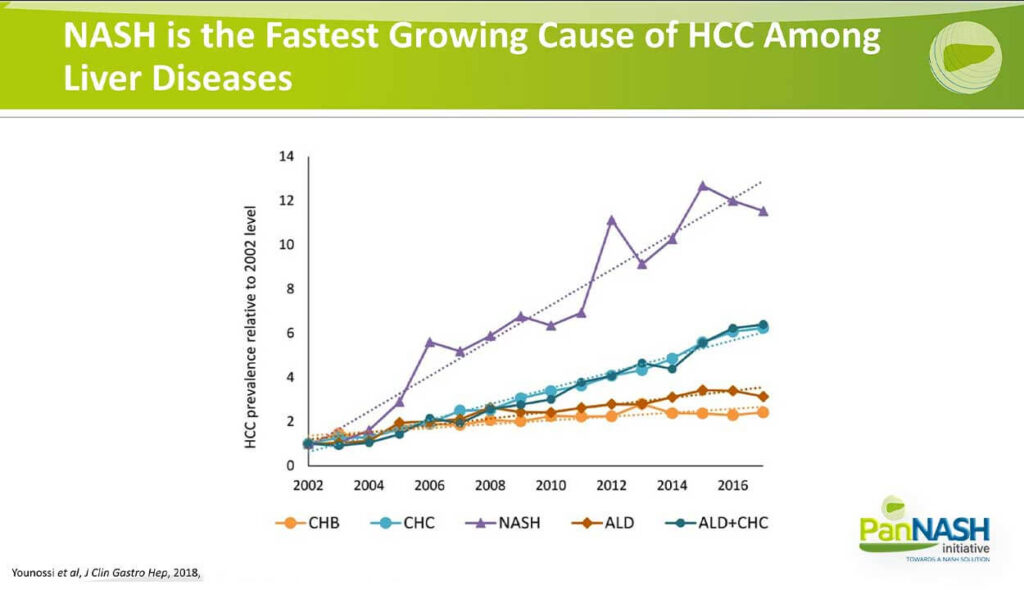
This slide illustrates the compelling epidemiologic data of HCC prevalence rising relative to the underlying etiology, including chronic hepatitis B, chronic hepatitis C, alcoholic liver disease or alcohol plus chronic hepatitis C. What is clear from the purple line is that relative to other underlying causes of HCC, the number or fraction of patients or relative rise associated with non-alcoholic fatty liver disease is disproportionately greater.
So over time, an increasing fraction of our patients with primary liver cancer or HCC will have underlying NAFLD or NASH, and this represents an important epidemiologic milestone that tells us we need to understand the disease better in order to intervene early and ultimately to develop more effective treatments.
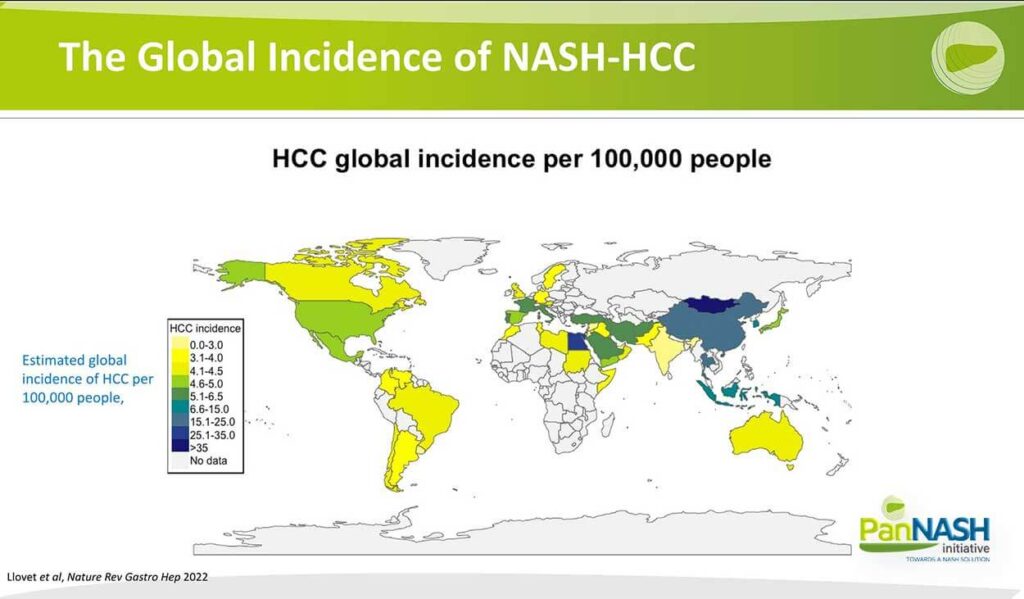
The Global Incidence of NASH-HCC
This slide from a recent review from my colleague Dr. Joseph Llovet underscores the global incidence of NASH HCC per hundred thousand people, with the darker blue representing the higher global incidence as one can see particular regions, including China and Mongolia, where co-infection with hepatitis B and delta is particularly high, but also the Middle East and North Africa in particular Egypt, so the disease is distributed differentially around the world but is increasing in all these regions.
> 1/3 of HCCs in NAFLD Occur in Non-Cirrhotics in a VA Population
One of the key points that I mentioned is that, unlike hepatitis C, D and other diseases, NAFLD as an underlying ideology is associated with a higher fraction of patients who are not yet cirrhotic. So what you’re looking at here is the relative distribution of cirrhosis in the darker grey and non-cirrhotic and a lighter grey again for hep C, B and alcohol. Almost over 90% in most cases of the cancers that arise do so after the patient at cirrhosis, whereas in NAFLD, up to a third of patients develop these cancers before they are cirrhotic, and this illustrates some important concepts.
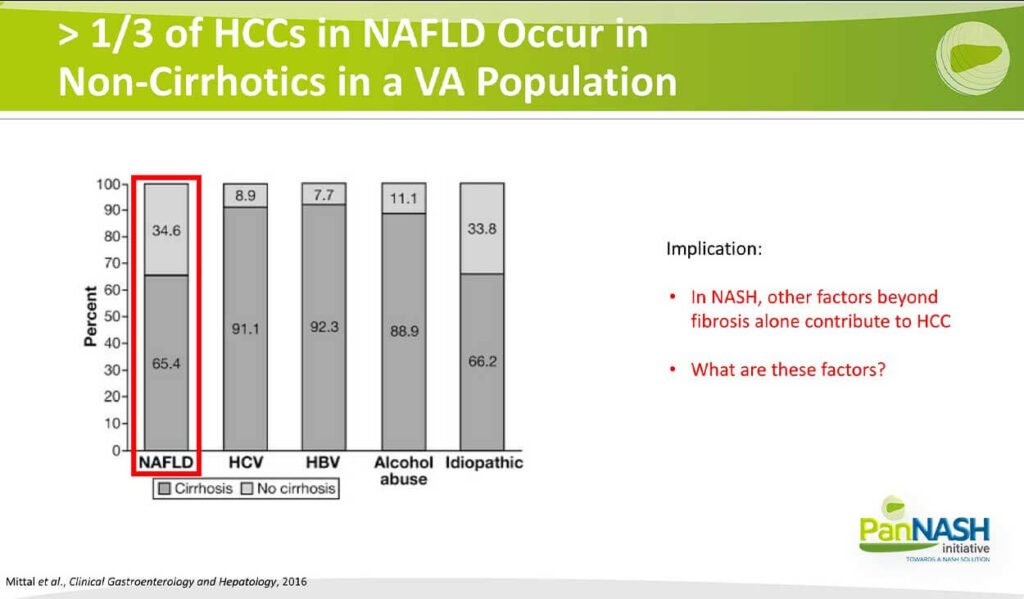
First of all, in NASH, it’s clear that other factors beyond fibrosis alone contribute to HCC, and It begs the question of what are these factors?
Cirrhosis is Still the Strongest Risk Factor for HCC in NAFLD
So before we focus on the factors apart from cirrhosis, it is worth reemphasizing that cirrhosis is still the strongest risk factor for HCC and NAFLD. Overall cirrhosis shown here as an incident rate is also linked to a higher incidence in males, which is well known to Hispanic ethnicity, and this is interesting because it’s almost surely related in part to the high prevalence of the polymorphism as I’ll describe in a moment. It is higher in Latin Americans.
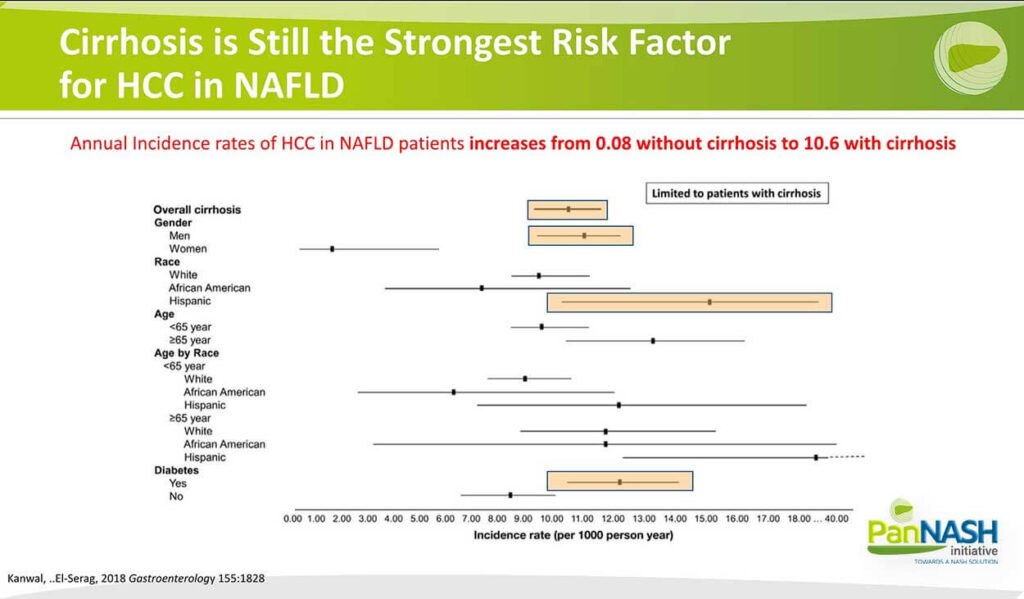
Also, the presence of diabetes is another heightened risk factor for cirrhosis in addition to hepatocellular carcinoma. So there are some identifiable risk factors epidemiologically, but also there are HCC mechanisms that are unique to NAFLD and not fibrosis dependent, and in particular, the prevalence of obesity in patients with NAFLD represents a significant risk factor for all cancers but in particular liver cancer.
HCC Mechanisms that are Unique to NAFLD or Non-Alcoholic Fatty Liver Disease and not Fibrosis-Dependant
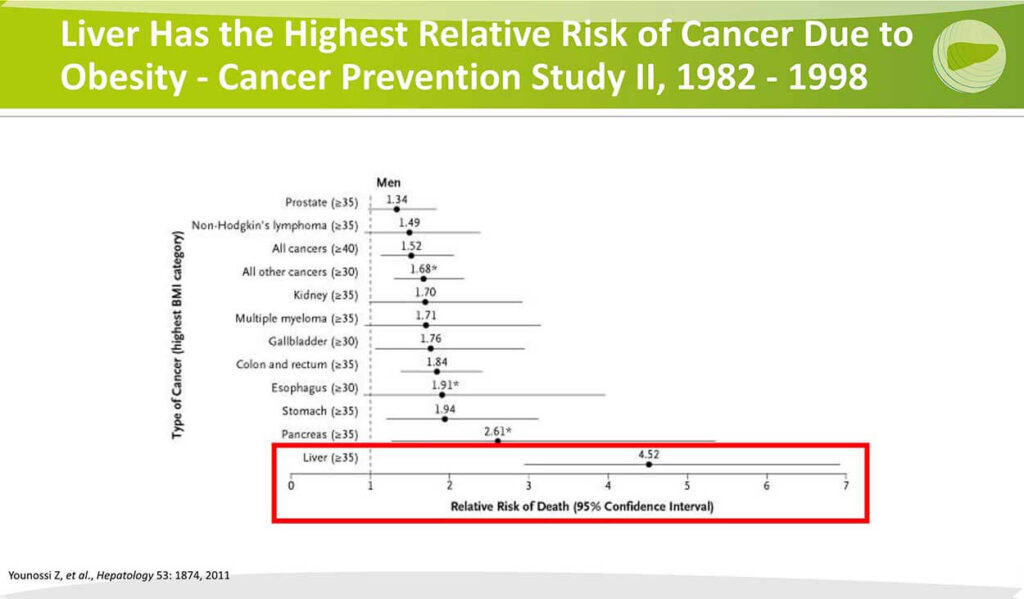
Livers Has the Highest Relative Risk of Cancer Due to Obesity
This is a study from over 10 years ago from DR Younossi looking at the type of cancer that’s impacted by the prevalence of obesity, and what you can see in the red box area is among the different cancers where obesity is a risk factor. That risk is heightened to the greatest extent for liver cancer. So clearly, obesity is creating some metabolic conditions that give rise to or predisposition to liver cancer, and of course, in some ways, it makes sense that that’s greatest in the liver compared to other organs because so much of the metabolic work of the system is and the storage of fat takes place in the liver.
Mechanisms of Increased Cancer Risk Associated with Obesity
There are other mechanisms of increased risk of cancer associated with obesity that are especially relevant to primary liver cancer. Obesity itself is a chronic inflammatory state with more oxidant stress, DNA damage and mutations, cause of chronic damage and DNA injury.
Secondly, with an increased fat burden in obesity. There is increased estrogen project production, and this may also drive the emergence of neoplasia more precipitously in the liver than in other organs; associated with this is a higher circulating level of insulin-like growth factor insulin, which are thought to be modestly pro-carcinogenic. More broadly, there’s an increase in all of the kinds, especially leptin, which is a mitogen and liver and so higher fat needs higher leptin, and higher leptin is a higher mitogenic signal. There are clearly genetic factors, which I’ll review In just a moment. And finally, we shouldn’t overlook the emerging importance of the altered gut microbiome.

It’s very clear that the microbiome is an important determinant both of disease and also of NASH in particular and HCC. Elegant studies in animals show that NASH itself can be transmissible through a gut microbiome transplant, and we know that patients who are at higher risk for cancer have a unique microbiome and the challenge now is to begin to harness the information from that microbiome both diagnostically, but also mechanistically to understand what it is about the features of the microbiome that predisposed to either NASH or cancer and to try to mitigate those therapeutically in addition to using it as a diagnostic tool.
So we have a lot to learn about the link between obesity, gut microbiome and these other features, but each of them represents potentially actionable points of pathogenesis that we have to understand better.
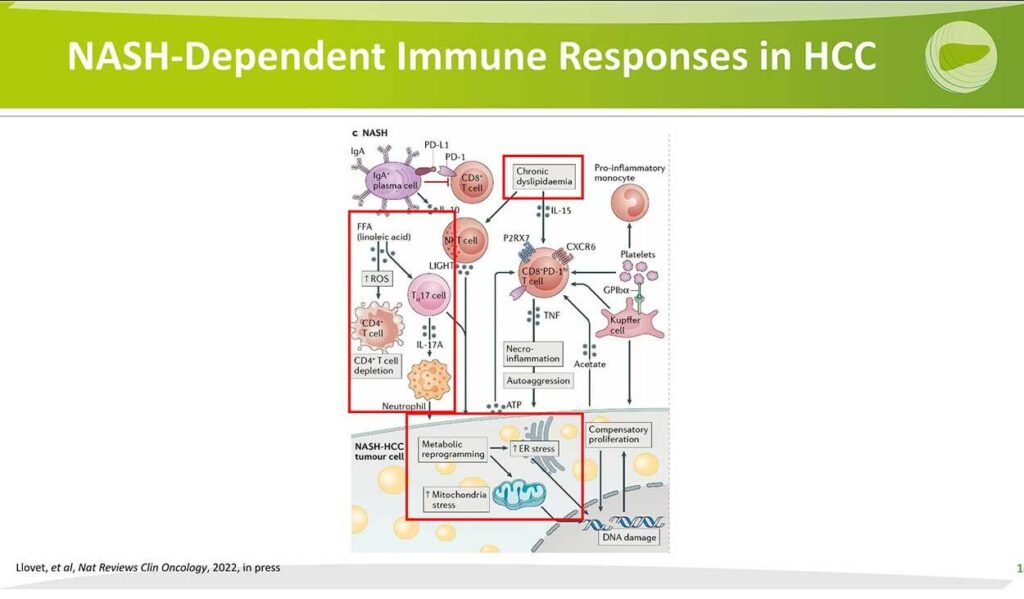
NASH-Dependent Immune Response in HCC
In addition, it’s very clear that the microenvironment in NASH is unique compared to other chronic liver diseases and creates a permissive microenvironment for cancer. This is summarized in another review by Dr Lovett that indicates the presence of chronic dyslipidemia, which ultimately, that can change the generation or increase the generation of reactive oxygen species, deplete protective CD4 cells and increase the 17 cells this can also lead to neutrophil activation and changes in this in the cytoplasm and nuclei and mitochondria, which collectively represents stress to the cell that may be unique to the NASH pathogenesis and all of these elements metabolic reprogramming increased in the plasmic reticulum stress and mitochondrial stress are thought they were stressors to the cell that can predispose to DNA damage, loss of protective abilities of the cell and ultimately the emergence of small cancer.
Article: NASH Limits Anti-Tumor Surveillance in Immunotherapy-Treated HCC
The importance of the microenvironment in NASH and the difference in this microenvironment compared to other etiologies was underscored by a very elegant paper published in Nature late last year, in which the investigators looked at the outcomes of treatment with the newly emerging immune checkpoint inhibitors and compared those outcomes between those patients who had NAFLD and those patients who had other etiologies almost highly viral hepatitis. You’re looking here at the overall survival curves in a discovery cohort in b and a validation cohort in c. In each case, the group with other etiologies had a better overall survival following checkpoint inhibitor treatment compared to the patients with underlying NAFLD, shown in blue both in the discovery and the validation cohort. So he suggests that there is a different microenvironment in the immune system in NAFLD compared to other radiology.
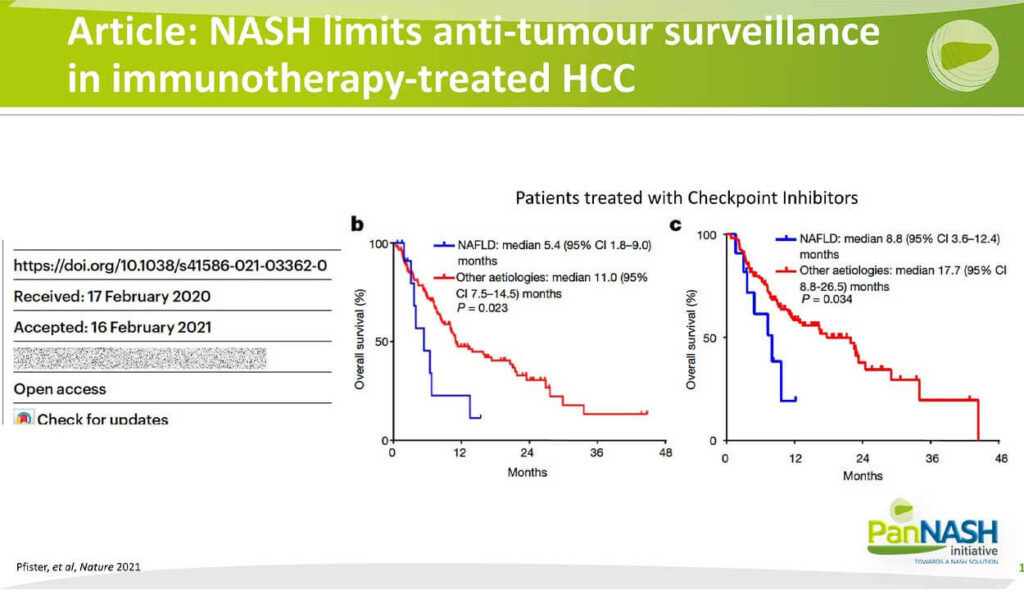
This is an area of intense interest and investigation. At the stage of this paper, the authors concluded that there might be NASH related to a Barren T Cell Activation that causes tissue damage and leads to impaired surveillance. So we are continuing to dig deeper into understanding the unique features of the immune microenvironment in hopes that we can harness that information to develop more personalized therapies that are NASH-specific and that have greater efficacy than the current immune checkpoint Inhibitors.
Genetic Contribution of PNPLA3 to NASH HCC
I also alluded earlier to the importance of genetics to incidents of NASH HCC, and I illustrate here data from some time ago from a group of Helen Reeves and the University of Newcastle looking at the rising incidents of HCC within their region in Northeast England, but more importantly the rising fraction of the overall number of cases that we’re attributable to NAFLD shown here in orange.
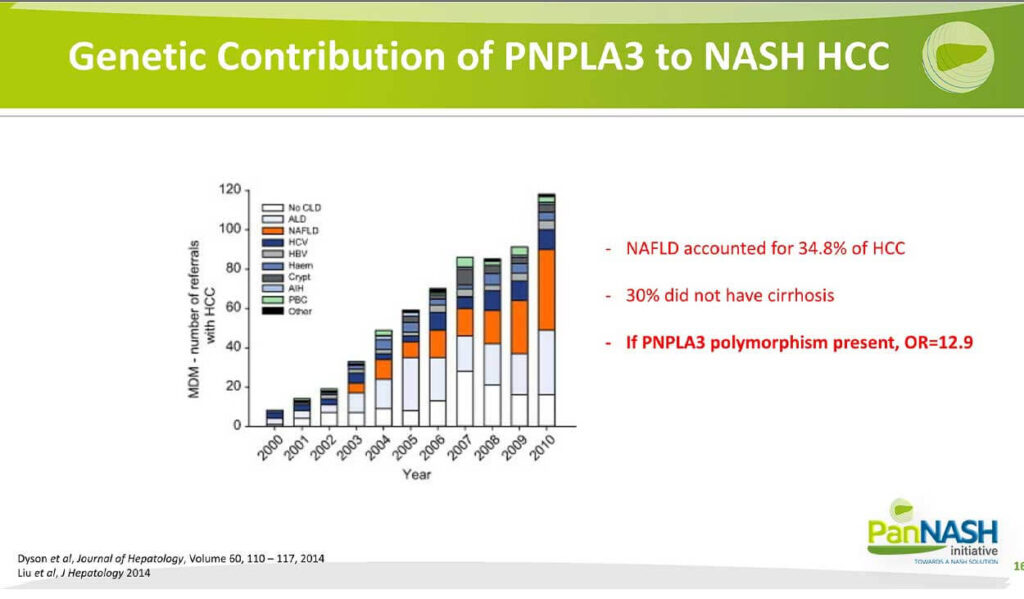
So what you can see overall is that not only was there an increase in the number of referrals which was expected, but also the overall fraction of those cases that had underlying NAFLD also increased and even at this 12 years ago now for the already accounted for almost 35% of their cancers. And again, the point that up to almost a third did not have ongoing cirrhosis, and this is a finding that’s been replicated now at least three times, the fraction of non-cirrhotic HCC is greater than other etiologies. But the key point I want to emphasize in this slide is if the risk-associated variant in this gene PNPLA3 was present, and particularly if it was homozygous, the odds ratio for the development of cancer was almost 13.
So it suggests again that polymorphisms in different genes, some of which are clustered ethnically or genetically and also predisposed to or create a setting right for the development of HCC.
Genetic Polymorphisms Linked To NASH-HCC
There are, in fact, a number of polymorphisms that are linked to NASH HCC – partially, as shown here also from another review by Dr Llovet. I’m not going to go through these individually, but I will point out that in virtually all cases, they relate to polymorphisms that somehow regulate or alter the metabolism of lipids, either export or synthesise content. And this suggests that changes in the amount of fat that’s accumulated based on genetic risks may also have downstream effects on the development of injury and also other factors related to fat accumulation that can drive the emergence of hepatocellular carcinoma.
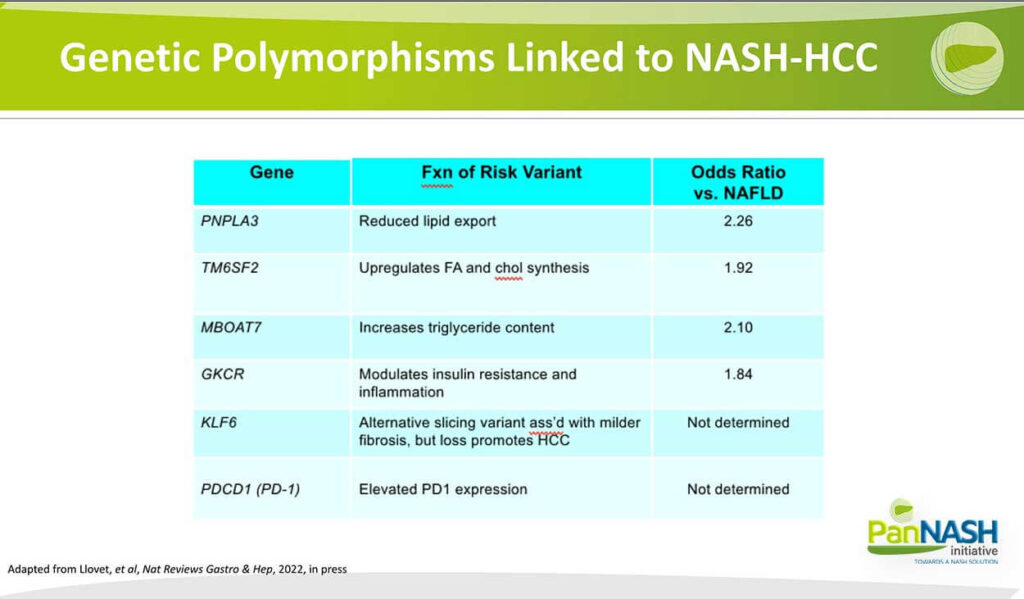
So over time will begin to integrate this genetic information to calculate more robust risk scores combined with other features of NASH to predict who will develop NASH HCC. This is important because it may improve our screening efforts so that patients who have a high genetic risk associated with liver fat could require more stringent and earlier screening for HCC to identify and treat cancers when they are still curable.
HCC Development in Western Diet/CCI4 FAT-NASH (“Fibrosis and Tumors”) Model
I’ll just say a word about experimental models that represent the mainstay overall for our thirst to understand NASH HCC. My own laboratory developed a model described here that we now call the fat NASH model. Fat stands for fibrosis and tumours, which we think is an important component of the model. These animals are given a high-fat, high-cholesterol, high-fructose diet with a very low dose of CCL4 carbon etrachloride.
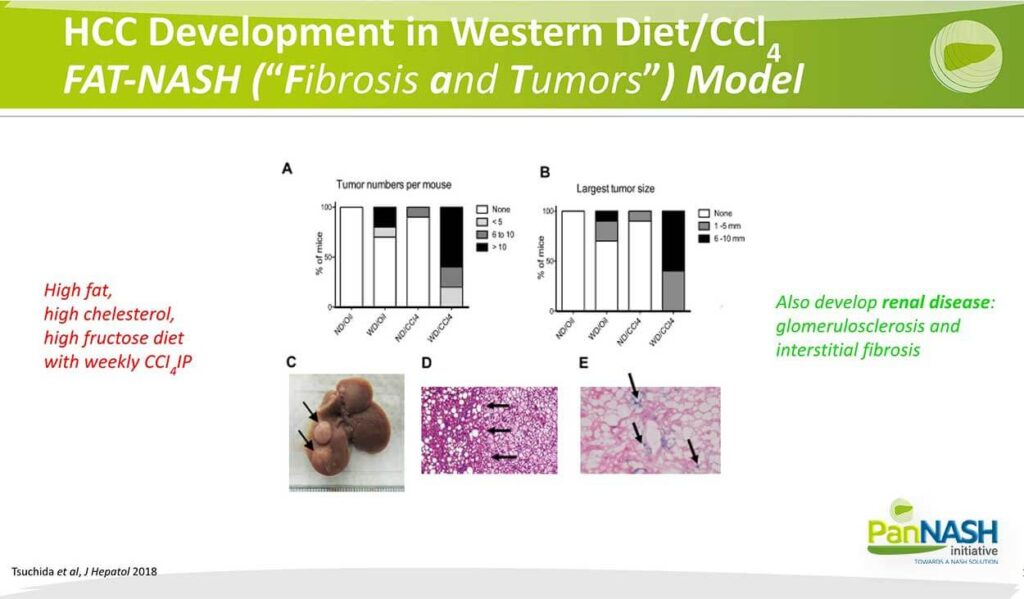
And what you can see is in the animals that receive both the western diet and CCL4, there was a dramatic increase in the number of tumours, and this means tumour size. Those tumours can be quite large and appear on the surface of the liver. Here you’re looking at a tumour liver interface with the NASH liver on the left and the tumour on the right. And studies that are still ongoing, we and others are exploring the possibility that, in the essence of cells indicated here by expression of beta-collector site Ace activity in blue, that’s in cirrhosis of sinusoidal cells in particular, which will review in a second, can ultimately represent some kind of risk for cholesterol.
I would also mention that what’s exciting about our fat NASH model is that, in addition to the liver disease, which we are studying extensively, we now have evidence that they develop a renal disease that includes glomerular sclerosis and interstitial fibrosis, and these are ongoing studies done in collaboration with our chief of nephrology here at Mount Sinai, but it does suggest that the model may be a useful one not only for liver disease but also for the metabolic syndrome, associated changes that occur in kidney and perhaps, in heart and blood vessels as well.
So let’s spend the last remaining few minutes talking about the presence of fibrosis and how that may impact NASH HCC.
Mechanisms Linking Fibrosis directly to NASH-HCC Hepatocellular Carcinoma
Molecular Differences between NASH-HCC and non-NASH-HCC
This is a very comprehensive table that illustrates the molecular differences between NASH HCC and non-NASH HCC. In each case. NASH HCC is on the left. So starting with the etiology, of course, Nash HCC is associated with obesity, diabetes, dyslipidemia, and metabolic syndrome, and the non-aged HCC is primarily attributable to underlying viral infection as well as alcohol.
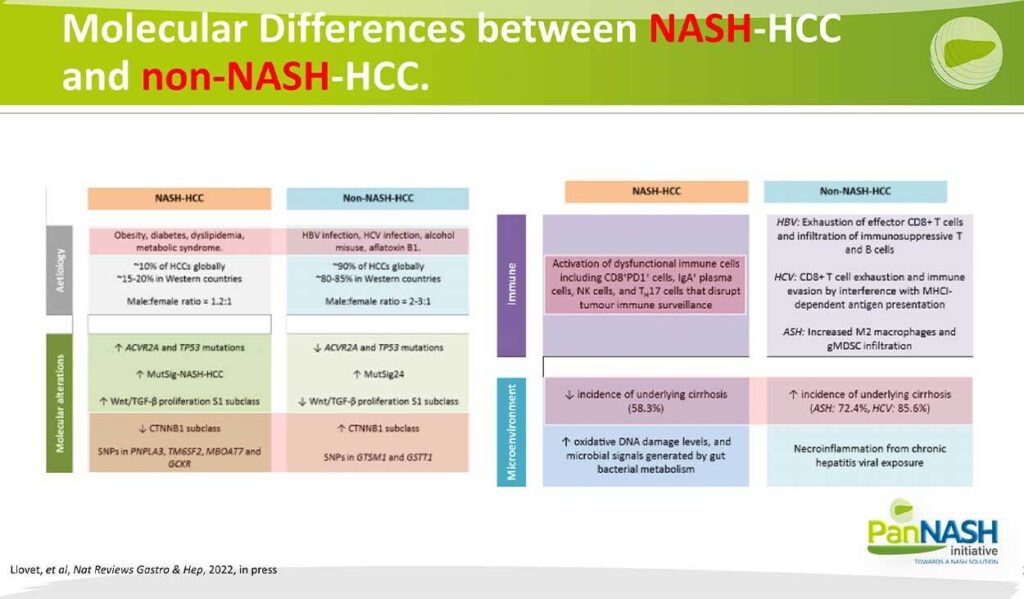
In terms of the molecular alterations, there’s been now extensive characterization suggesting that in NASH HCC, a smaller fraction of the tumours had dysregulation of the wind beta cotinine subclass, which is deviated CTN and B1. Whereas non-nash HCC has a higher fraction of tumours that have dysregulation of wind beta Katina. The snips that I described in the previous slide are also more prevalent in NASH HCC, the non-NASH HCC. In terms of the immune phenotype, I’ve already emphasized that we think this is quite different in the underlying NASH that gives rise to HCC, and that includes many of these features here of activated dysfunctional immune cells as well as in case cells and elevated TH17 cells that disrupt tumour surveillance.
Again, the key point is that there’s a decreased incidence of underlying cirrhosis in Nash HCC there to non-NASH HCC. Interestingly NASH in more recent studies also has a higher fraction of non-cirrhotic HDC, but not as high as those who have NASH. Also, remember that presence of NASH and Ash together is not uncommon. So presumably, this is a synergistic interaction that accelerates the emergence of both NASH, also inflammation fibrosis and cancer when both NASH and ASH are present in the same patient.
Hepatic Stellate Cell Activation – A Central Event in Liver Fibrosis
As many may know, my own laboratory is focused for almost 40 years on the pathogenesis of NASH or fibrosis. I should say and in particular, the role of the hepatic stellate cell; the hepatic stellate still shown here in blue, is a resident non-parenchymal non-hepatocyte cellular liver located between hepatocytes and sinusoidal endothelial cells. It is, in fact, a parasite of the liver that wraps its foot processes around the sinusoid.
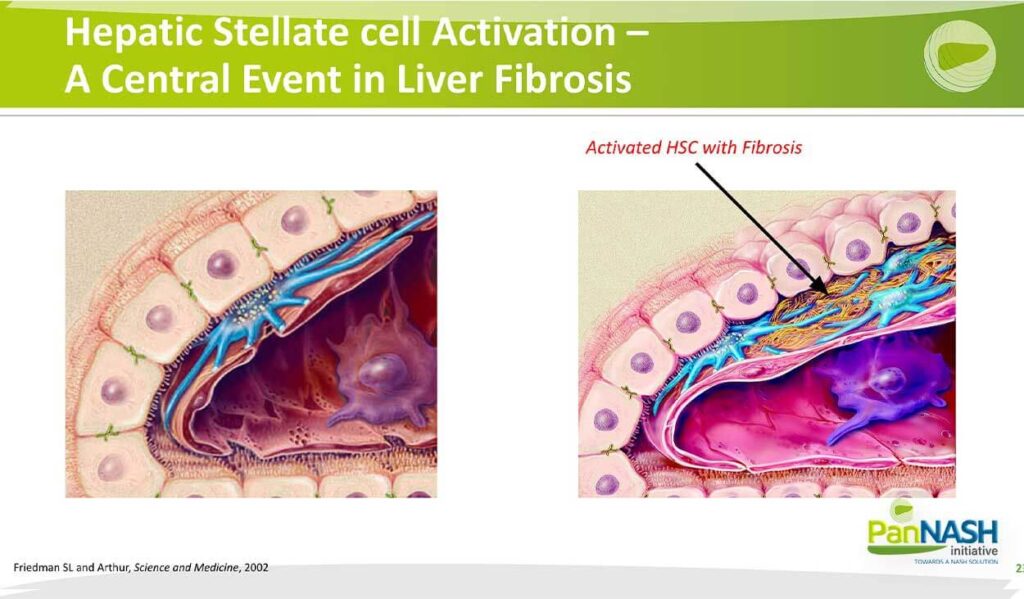
And its most characteristic feature is the storage of droplets of vitamin A in the form of retinol esters. As the cells activate shown here, those retinal esters are lost and the cells begin to multiply, associated with accumulation of scar or interstitial collagen and resulting in changes in the function of surrounding cells, in particular loss of hepatocyte differentiation, closure of the pores in the subendothelial cells or finasteride that normally constitute a healthy sinusoid and activation of macrophages.
So our studies have focused for decades on how activation occurs and, of course, how It ultimately can be mitigated to reduce fibrosis therapeutically.
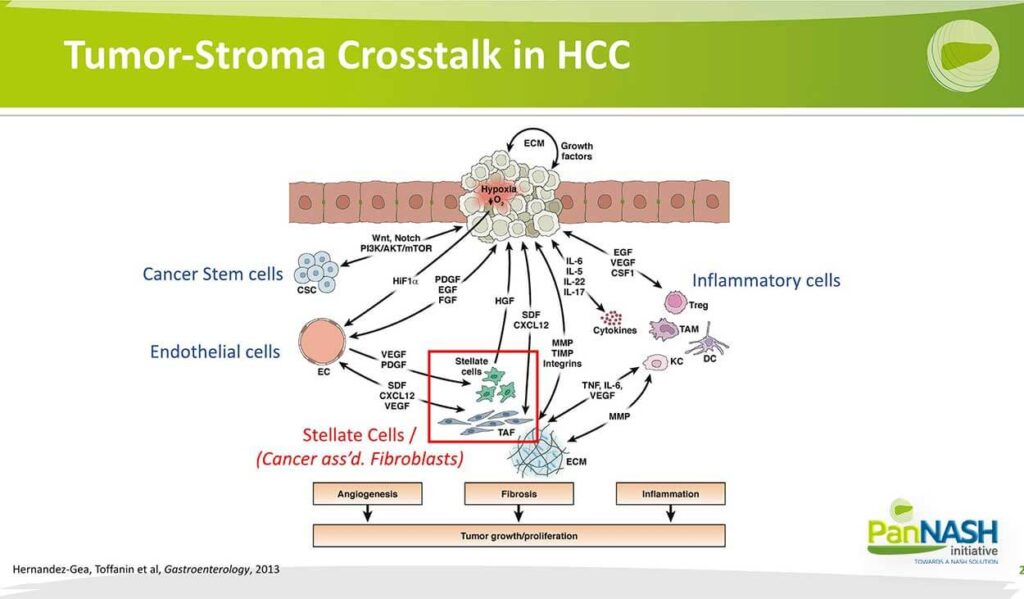
Tumor-Stroma Crosstalk in HCC
The role of these stellate cells in tumour emergence, both primary and probably metastatic tumours, is multifold stellate cells, which also are known in cancer-associated or tumour-associated fibroblasts caffs or talfs play an essential role in integrating signals both from damage to a pedicides that include hypoxia. But also interactions with endothelial cells as well as different immune cells subsets and collect totally stellate cells that help orchestrate both the angiogenic fibrotic as well as the inflammatory responses that underlie tumour growth and proliferation.
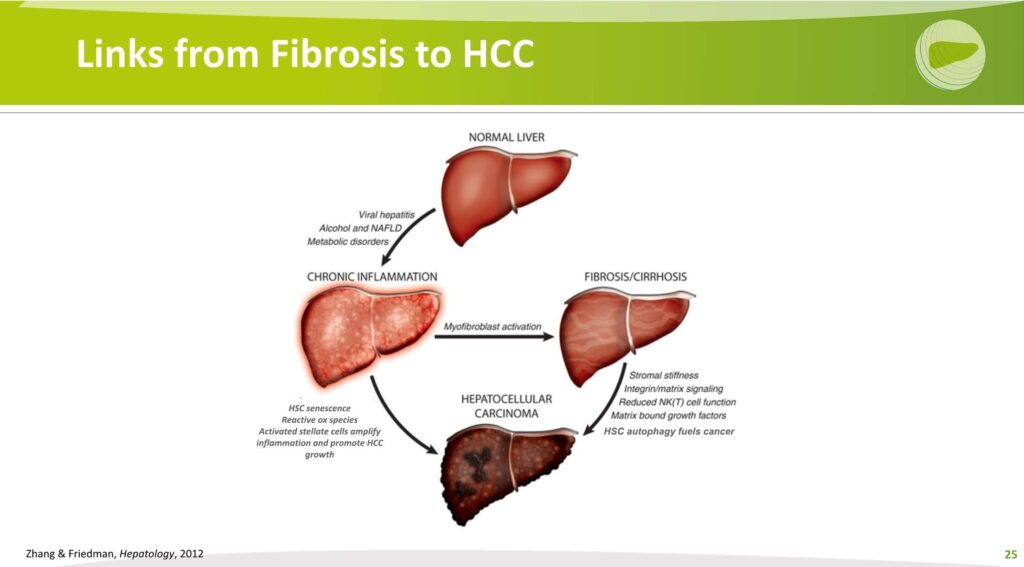
Links from Fibrosis to HCC
We summarized some time ago the potential pathways and links between fibrosis and HCC. I refer you to this reference from 10 years ago, which fundamentally is still accurate in terms of the concepts we outlined. Time does not permit me to go through all of these, but there are certain pathways that go from normal to chronic inflammation. Other pathways that underlie the emergence of hepatocellular carcinoma directly from the inflammatory liver, but also through myofibroblast or stellate cell activation that generate fibrosis and lead to a number of other features that also contribute.
Let me just illustrate one or two of these: for one thing, it turns out that stellate cells, as I mentioned, have a nodal point regulating immunology immune responses and inflammation in a way that may promote HCC growth. Summarized in this slide here where you’re looking at an activated stellate cell and all of the different ways that stellate cells can amplify inflammation.
I’ll emphasize for the purpose of this talk there are a couple of specific pathways that are emerging; in particular, that stellate cells can enhance Myeloid-derived suppressor cells and suppress T and B cells via PDL, one of which is expressed on stellate cells. And this effectively can lead to a more immunotolerant environment so that the immune system does not attack and clear national tumours. So in a sense, the immune tolerance of the liver, which is largely attributable to the activation of stellate cells, also creates a permissive environment in which the immune system can no longer surveil the tumours or surveil the tissues to accurately drive the immune system to clear early tumours.
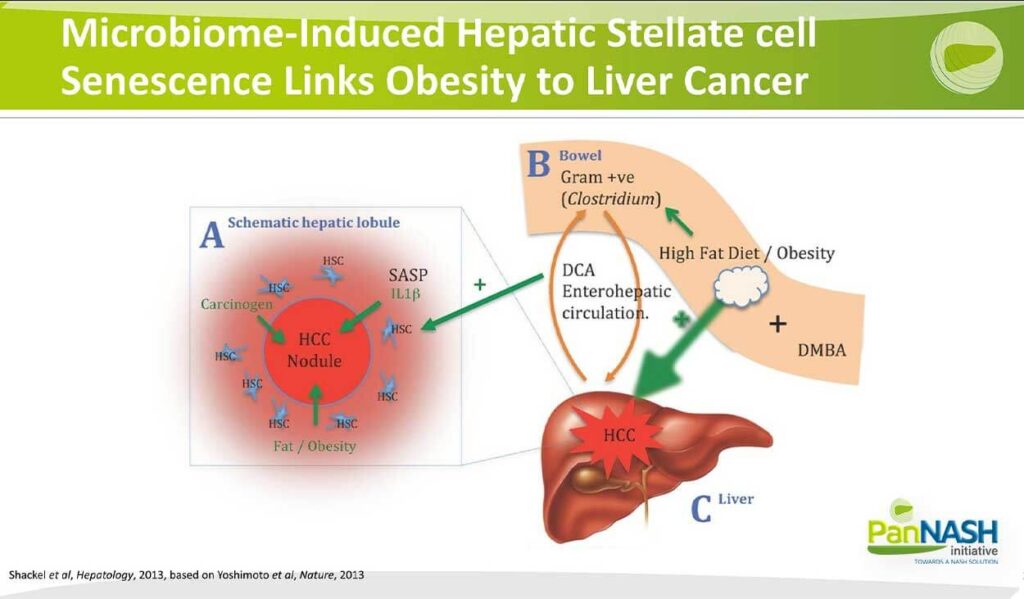
Microbiome-Induced Hepatic Stellate Cell Senescence Links Obesity to Liver Cancer
I also mentioned earlier the importance or the emerging importance of a senescence-associated secretory phenotype or SAS. Several studies done years ago underscored this potential role specifically of stellate cells becoming senescent and driving the development of a pro-thermogenic environment. This is linked in at least in one study from Japan to a high-fat diet, a change in the microbiome and an elaboration of the oxalic acid that drives the senescent microenvironment at the liver. So this is an exciting potential area of interaction where the gut changes in the microbiome lead to changes in the senescence microenvironment that develop into liver cancers.
Anti-uPAR CAR T cells Reduce Fibrosis
In one exciting study performed by the laboratory of Dr Scott Lowe, we participated peripherally in this study in which, the reason that if they could clear the senescent stellate cells, then perhaps that would have an impact on fibrosis. And they did this by generating specialized, focused CAR T or chimeric antigen receptor T cells that were conditioned or generated to identify a very specific target that’s only expressed on stellate cells…
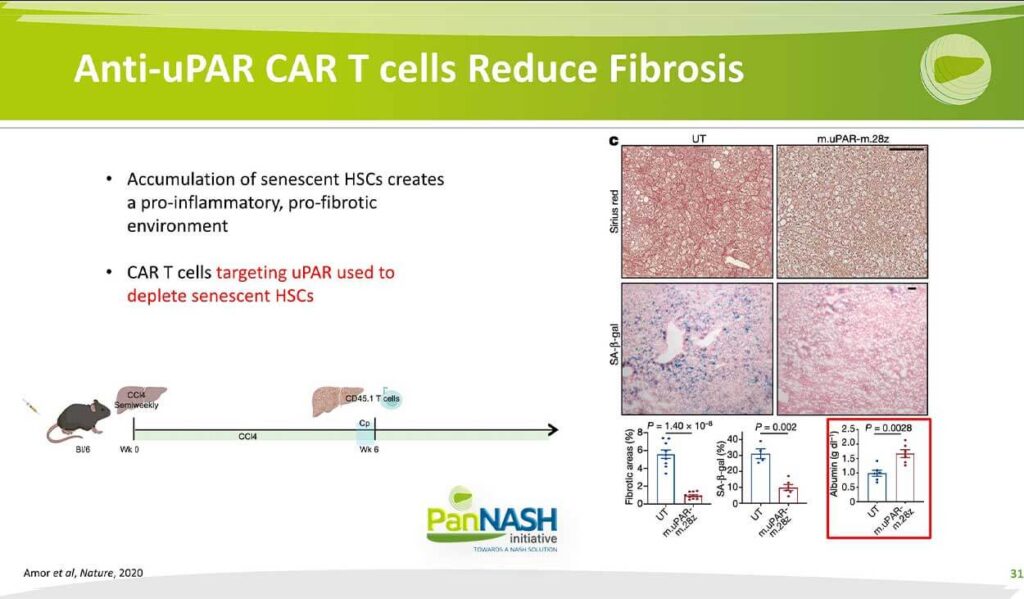
In this study published in nature two years ago, they gave CCL4 to develop fibrosis. They’ve also done this with a NASH model at six weeks into the injury. They administered very specialized T cells to kill any stellate cell that was senescent… And these results were quite striking when they cleared senescent stellate cells o the right; based on serious red staining, there was a dramatic demolition and fibrosis content quantified here in the lower left. In addition, when they used beta-gal activity staining to identify senescent cellular cells, it was a dramatic diminution in beta-gal expressing cells suggesting that, indeed, their car T cells effectively or successfully cleared those synesthetic cells, but they also showed what I find a particularly interesting and exciting is that as they clear, it’s, in essence, stellate cells liver function improved and serum albumin almost doubled. And this suggests an important link that we don’t understand well between fibrosis and regeneration, the more fibrosis, the less regeneration. In this case, once you clear senescent profibrotic stellate cells, regeneration may be Amplified, and that’s reflected in the rise in serum albumin in these animals.
Links from Fibrosis to HCC
In addition, although I won’t illustrate it for lack of time, selling cells and hepatocytes in the microenvironment can interact with matrix-bound growth factors as the scar accumulates. Several growth factors are Tethered to that scar and can be released as Stellate Souls activate further and ultimately drive replication and proliferation of hepatocytes and the emergence of cancer. So there are both immune but also physical and cytokine-driven interactions between stellate cells and the environment they create and the emergence of liver cancer hepatocytes.
NASH-HCC+ Fibrosis – Summary
So this gives me an opportunity to summarize the key points I’ve been trying to make it for the last few minutes. First of all, the systemic effects of obesity on cancer development are especially relevant to the liver and involve inflammation, genetics, growth factors out of a kind and microbiome, among others, and I gave you several illustrations.
The immune milieu of NASH is distinct and may be immunotolerant and thereby diminishing responses to checkpoint Inhibitors. So work needs to be done on defining the specific immune deficits in the NASH liver so that we can establish more personalized or precision therapies that attack the immune dysregulation that’s unique to NASH.
Finally, mechanisms linking fibrosis to HCC and NASH include actions of the hepatic stellate cells as modulators and orchestrators of immunity and inflammation, but also the physical features of the extracellular matrix, their ability to find growth factors and also the potential stiffness of the liver that occurs as fibrosis progresses.
The last point is that as we develop and approve NASH therapies and antibiotic treatments, we should explore their impact on these competitive mechanisms of HCC in the hope I expect that the therapy will not only improve and reduce the risk of liver failure but will also attenuate the emergence of liver cancer.
So with that, I’ll close. Thank to the PanNASH Initiative for the opportunity to present this lecture, which I hope you find informative. Thank you.



