
Overview of Pathogenesis of Nonalcoholic Steatohepatitis
An understanding of the pathogenesis and natural course of NAFL and NASH is essential. These conditions are not static, but dynamic, and may progress or regress at variable rates in different individuals or even in the same individual at different times.
Non-alcoholic steatohepatitis (NASH), resulting from a combination of adipose tissue insulin resistance, adipocytokine imbalance and systemic inflammation, is currently a major worldwide cause of chronic liver disease, contributing to cirrhotic morbidity, hepatocellular carcinoma and liver transplantation, and worsening cardiovascular disease and metabolic dysfunction.
RELATED Pathophysiology of NASH VIDEOS

Clinical Chemistry In NAFLD Part 3 – Biomarkers and Hepatic Fibrosis
Discover the intricate dynamics of Clinical Chemistry in NAFLD with Dr. Guillaume Grzych, a leading

Clinical Chemistry in NAFLD Part 2 – Biomarkers, Hepatic Steatosis and NASH
Discover how key biomarkers like Fatty Liver Index and SteatoTest are transforming NAFLD diagnosis and

Key Messages from 2 Recent Studies on NAFLD Severity in People with Type 2 Diabetes
Unveiling the high prevalence of NAFLD and advanced fibrosis in type 2 diabetes patients –

NAFLD Pathophysiologic Role of Intrahepatic Vasculature
Dr. Kwanten (Belgium) discusses the pathophysiological role of intrahepatic vasculature in non-alcoholic fatty liver disease
RELATED Pathophysiology ARTICLES
Hepatic inflammatory responses in liver fibrosis
Recent technological advancements have provided unprecedented insights into the inflammatory mechanisms underlying liver fibrosis, leading
Exploring the Promise of Pan-PPAR Agonism
Peroxisome proliferated-activated receptors (PPARs), including α, β/δ and γ isotypes, are a nuclear receptor family
Managing Hepatocellular Carcinoma in NAFLD: a review
Currently, there are rising levels of non-alcoholic fatty liver disease (NAFLD). With this, comes a
The new paradigm of cardiometabolic syndrome: a review
Cardiometabolic syndrome (CMS) involves a complex interplay of many issues, involving obesity, metabolic dysregulation, cardiovascular
Environmental and genetic factors promote NASH pathogenesis
1.Diehl AM, Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med. 2017;377(21):2063-72.
2. Haas JT, Francque S, Staels B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu Rev Physiol. 2016;78:181-205.
3.Naik A, Kosir R, Rozman D. Genomic aspects of NAFLD pathogenesis. Genomics. 2013;102(2):84-95.
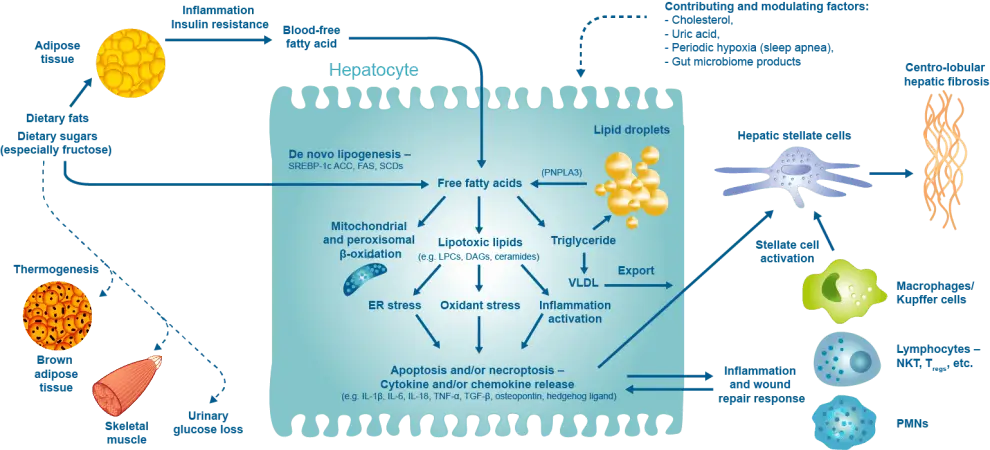
Lipotoxicity
Hepatic inflammation is an important component of the process, but it is unclear whether it is a primary cause or consequence (or both) of hepatocyte injury and death.
1.Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci U S A. 1994;91(23):10878-82.
2.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52(2):774-88.
3.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908-22.
4.Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144(12):5159-65.
5.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. Jama. 1999;282(17):1659-64.
6.Liss KH, Finck BN. PPARs and nonalcoholic fatty liver disease. Biochimie. 2017;136:65-74.
The inflammatory and immune systems
1.Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142(4):711-25
2. Barb D, Portillo-Sanchez P, Cusi K. Pharmacological management of nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1183-95
Oxidative stress and mitochondrial dysfunction
1.Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid Med Cell Longev. 2018;2018:9547613.
2. Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol Commun. 2018;2(12):1425-39.
3.Jelenik T, Kaul K, Sequaris G, Flogel U, Phielix E, Kotzka J, et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes. 2017;66(8):2241-53.
4.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21(5):739-46.
5.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18(6):716-31.
6.Wei Y, Wang D, Gentile CL, Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem. 2009;331(1-2):31-40
Inflammation
Liver inflammation in NAFLD can be triggered outside the liver such as in adipose tissue and the gut as well as inside the liver. Increased visceral adipose tissue is associated with increased infiltration of inflammatory macrophages, which triggers insulin resistance and inflammation in the adipose tissue and leads to a disturbed adipokine profile, namely high leptin and tumour necrosis factor (TNF) levels and low adiponectin levels. While adiponectin reduces insulin resistance, liver steatosis and inflammation, TNF increases insulin resistance and is pro-inflammatory.
1.Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. 2018;15(6):349-64.
2. Buechler C, Wanninger J, Neumeier M. Adiponectin, a key adipokine in obesity related liver diseases. World J Gastroenterol. 2011;17(23):2801-11.
3.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40(1):46-54.
4.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver. 2012;6(2):149-71.
5.Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(1 Suppl):S47-64.
6.Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A. 2014;111(26):9597-602.
7.Krenkel O, Tacke F. Macrophages in nonalcoholic fatty liver disease: a role model of pathogenic immunometabolism. Semin Liver Dis. 2017;37(3):189-97.
Fibrosis
1.Diehl AM, Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med. 2017;377(21):2063-72.
2. Haas JT, Francque S, Staels B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu Rev Physiol. 2016;78:181-205
3.Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu Rev Pathol. 2016;11:451-96.
4.Angulo P, Machado MV, Diehl AM. Fibrosis in nonalcoholic Fatty liver disease: mechanisms and clinical implications. Semin Liver Dis. 2015;35(2):132-45.
5.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028-40.
6.Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13(22):3056-62.
7.Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, et al. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol. 1999;276(4):G1059-68.
8.Dudás J, Kovalszky I, Gallai M, Nagy JO, Schaff Z, Knittel T, et al. Expression of decorin, transforming growth factor-beta 1, tissue inhibitor metalloproteinase 1 and 2, and type IV collagenases in chronic hepatitis. Am J Clin Pathol. 2001;115(5):725-35.
9.Xu F, Liu C, Zhou D, Zhang L. TGF-beta/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64(3):157-67.
10.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10(1):76-99.
- Anstee QM, Mantovani A, Tilg H, Targher G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2018;15:425-39.
- Armandi A, Schattenberg JM. Beyond the paradigm of weight loss in non-alcoholic fatty liver disease: from pathophysiology to novel dietary approaches. Nutrients. 2021 Jun 8;13(6):1977.
- Francque SM, van der Graaff D, Kwanten WJ. Non-alcoholic fatty liver disease and cardiovascular risk: pathophysiological mechanisms and implications. J Hepatol 2016;65:425-43.
- Lonardo A, Mantovani A, Lugari S, Targher G. Epidemiology and pathophysiology of the association between NAFLD and metabolically healthy or metabolically unhealthy obesity. Ann Hepatol. 2020 Jul-Aug;19(4):359-366.
- Sanyal A. Genetics of nonalcoholic steatohepatitis. Gastroenterol Hepatol (N Y). 2020 Dec;16(12):651-653.
- Steinman JB, Salomao MA, Pajvani UB. Zonation in NASH – A key paradigm for understanding pathophysiology and clinical outcomes. Liver Int. 2021 Jul 30 [Epub ahead of print].
- Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020 Sep;69(9):1691-1705.
- Tilg H, Effenberger M. From NAFLD to MAFLD: when pathophysiology succeeds. Nat Rev Gastroenterol Hepatol. 2020 Jul;17(7):387-388.
- van der Graaff D, Kwanten WJ, Francque SM. The potential role of vascular alterations and subsequent impaired liver blood flow and hepatic hypoxia in the pathophysiology of non-alcoholic steatohepatitis. Med Hypotheses. 2019 Jan;122:188-197.
- Vonghia L, Michielsen P, Francque S. Immunological mechanisms in the pathophysiology of non-alcoholic steatohepatitis. Int J Mol Sci. 2013 Oct 1;14(10):19867-90.
- Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11-20.