INTRODUCTION ABOUT Dr. Wilhelmus Kwanten
Hello everyone; I’m Wilhelmus Kwanten. I’m a research physician in Antwerp working at the Antwerp University Hospital. My research interests are in the vascular alterations related to NAFLD, being the topic of this talk.
OVERVIEW OF NAFLD Pathophysiology Role Of Intrahepatic Vasculature
Here’s the overview of my lecture. I will cover multiple areas to talk about the pathophysiological role of this vasculature intrahepatically in NAFLD:
Table of Contents
- Early NAFLD Leads To PHT
- Higher PoHT in NAFLD?
- Portal Hypertension
- The Antwerp Hypothesis
- Ultrastructural Changes
- Changes in Portal Vasculature
- LSEC Capillarisation
- Vascular Dysfunction – IHVR
- Vascular Dysfunction – Vasodilatation (NO)
- Vascular Dysfunction – Vasoconstriction (Mx and ET-1)
- Vascular Dysfunction – Bosentan
- Structural and Functional Alterations – Summary
- Liver Tissue Hypoxia
- Upcoming Drugs
- Take Home Messages
EARLY NAFLD LEADS TO PHT
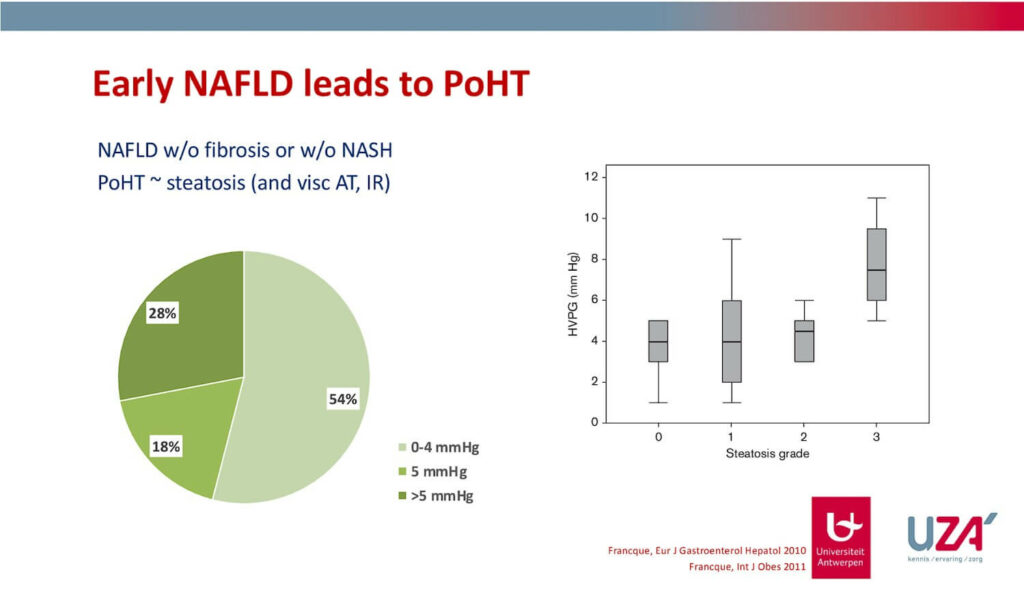
We can already see portal hypertension in the early stages of NAFLD in about half of the patients. The majority of these patients are not even faced with fibrosis or steatohepatitis.
Diving deeper into this data, we can see that there is an association between the visceral adipose tissue, insulin resistance, as well as the degree of steatosis leading to a certain degree of portal hypertension.
Findings of other groups recently confirmed our observations, showing that you can already see some portal hypertension even if cirrhosis still isn’t present.
Cirrhosis is faced with clinically significant portal hypertension exceeding 10 or even 12 millimeters. This was only the case in one non-serotic NAFLD patient.
But the most important observation is that 17% of these patients with an F0 to F3 can already be seen with some degree of portal hypertension.
Higher PHT in NAFLD?
Portal hypertension in NAFLD isn’t as straightforward as other liver diseases. What we are now looking into is not early NAFLD but a really enhanced hypertension by NAFLD with cirrhosis. We can see that the measured or the estimated portal degree of portal hypertension by HVPG is more often discongruent with the real portal pressure, being underestimated.
When looking into a cohort of patients that got TIPSS placed and looking at a larger cross-sectional study comparing NAFLD with viral hepatitis, we can see that the degree of compensation is higher at lower levels of HVPG. This implies that the effective portal hypertension might be even a little bit higher than estimated by the HVPG technique.
Portal Hypertension
Zooming into the background of portal hypertension – we are all aware of the fact that this is the consequence of either the increased intrahepatic vascular resistance being the result of both structural as well as functional alterations, as well as an increase in the flow of the splanchnic veins that are vasodilated.
Collaterals or shunts might even decrease the PHT potential for a certain amount but might not fully relieve the portal hypertension.
These structural and functional alterations appear to be quite important when talking about NAFLD, which I will explain in the next slides.
The Antwerp Hypothesis
For approximately more than 10 years we believe that the central driving force of NAFLD to NASH are the intrahepatic vascular alterations that can be found early on in early NAFLD.
These vascular alterations do not only lead to an increased intrahepatic vascular resistance with consequent portal hypertension but also might induce local tissue hypoxia (driving force of this progression).
Intrahepatic resistance is central and key to NAFLD, at least in our eyes, as it leads to portal hypertension (explained a little bit earlier on). It also leads to a decreased blood supply with subsequent hypoxia to deliver tissue, partially relieved by increased arterial blood flow by the buffer response, but this is a driving force.
Structural and functional changes (examples will be given shortly) are for instance the narrowing of sinusoids or distortion of the architecture of the vasculature or either vascular dysfunction leading to a hypo-constrictive net effect in the liver.
Ultrastructural Changes
Here are some examples of these changes on the structural level showing normal levels in steatosis or animal models of steatosis (on top of this slide). The normal architecture is quite distorted; the regular organization has disappeared. We also see more blunt endings and some BLAPs on the sinusoids.
This is repeated in different studies with different models of steatosis, for example in an MCD diet, where you can again see a distortion in the architecture and a much lower sinusoid quality.
Changes in Portal Vasculature
Other studies looked into the structural organization of nerves and veins within the liver. They could show that when looking into normal livers compared to livers affected by steatosis (panel P and D), the central veins (in gray) are not that affected. The portal veins (shown on the right and in the far right panel), have a real decrease in area and in volume, showing that they are compressed and are structurally altered in NAFLD.
LSEC Capillarisation
The CD34 stain (human data) is present in lower degrees of fibrosis and is related to the capillarization of the sinusoids. You can see that by increasing the degrees of fibrosis or inflammation, the marker of capillarization (structurally altered sinusoids), gradually increases.
Vascular Dysfunction – IHVR
Moving into functional altercations, we can now see that the vascular function is disturbed by PoHT hypertension (not shown on slides). By looking into the intrahepatic vascular resistance you can see that it is also increased over a wide range of lows.
Vascular Dysfunction – Vasodilatation (NO)
The intrahepatic vascular resistance is the result of the diminished phase of the dilatation as has been proven in different models where you can see that the acetylcholine-mediated vasodilation working through the release of nitrogen monoxide fire at the endothelial cells which is blunted when you have NAFLD present.
Vascular Dysfunction – Vasoconstriction (Mx and ET-1)
On the other hand, the opposite vasoconstrictive reaction is altered but is more severe due to an increase in vascular resistance. So the response to methoxamine, which is an α-1- Agonist or endothelium, is increased in steatotic animals compared to controls.
Vascular Dysfunction – Bosentan
On the right you have some graphs on endothelin 1. When we block endothelin 1 with Bosentan, which is an antagonist of the receptor, we can see that the pressure is actually back to normal in animal modus of either NAFLD or NASH.
In line with normalization of the intrahepatic vascular resistance, we can see – an improvement of liver biochemistry and a small but noticeable improvement of the level of steatosis (not only functionally but also structurally) where we can see that the distorted architecture is nearly normalized when treating with both Bosentan.
Structural and Functional Alterations – Summary
I only had the time to give some examples, but there are many more involved and already observed to be changed in early NAFLD. I did not speak a lot about sclerotic NAFLD, but it’s commonly believed that whilst the vascular alterations are present in early NAFLD and quite specific to NAFLD, the further the disease progresses and the most cirrhosis you have, the more similar it becomes to cirrhosis of other origins.
There might be pre-sinusoidal components in NAFLD which is concluded by the observed underestimation of the PoHT pressure, as discussed a little bit earlier.
Liver Tissue Hypoxia
Do these alterations indeed lead to hypoxia as we hypothesized?
Yes, we think so, and we can prove it. If we look at this slide, we can measure the oxygen tensions directly in the livers of stereotic animals showing that the oxygen tension has decreased. But also, when we stand for Pimonidazole, which is a marker of hypoxia, we can visually see that the amount of stain is increased, but when we quantify this when correcting for the amount of steatosis present, we can see that this is markedly increased mostly in the PoHT regions. So hypoxia is indeed present.
Upcoming Drugs
Functional alterations means that you might be able to modulate these alterations in a way in which we can hold or improve the disease. This is a list of certain drugs that are currently quite far in the pipeline (on the slide), but many of them do not have any data on PoHT hypertension intrahepatic vascular resistance. Only obeticholic acid and Lanifibranor have some data.
Obeticholic Acid (OCA) – FXR agonist
Obeticholic acid is mostly studied in models of cirrhosis where if you have cirrhosis induced by TAA or BDL for instance and you add obeticholic acid, the intrahepatic vascular resistance as well as the pressure is decreased and not normalized after treatment.
This was also seen in models of prehepatic PoHT hypertension by PPvl or by CCL4 where the obeticholic acid but also other effects are agonists and are able to reduce the pressure. So it seems to be a class effect.
It’s mostly believed to be brought along by a decrease in the intrahepatic vascular resistance as the PoHT pressure decreases but the inflow of blood does not change, meaning that it has to be related to the vascular alterations at the level of the liver.
Lanifibranor (panPPAR agonist)
The panPPAR agonist is closely woven with all the different components of the sinusoids, and it is not surprising that they might have an effect on PoHT hypertension as well as the Intrahepatic vascular resistance.
There is some preliminary data in our lab that shows that NAFLD might work, but I cannot present this at the present time. But, by looking into models of enhanced disease, we can show that PoHT hypertension, which had increased, was decreased by giving Lanifibranor. The effect of acetylcholine, the nitrogen monoxide-derived vasodilation, was also improved after giving Lanifibranor, meaning that this might help on an ultrastructural level. There is also some improvement observed in the TAA model.
Take Home Messages
So in this short but dense talk with a lot of information, I hope I could convince you all that PoHT hypertension might already be present in patients with NAFLD and that this is an early phenomenon.
These intrahepatic vascular alterations leading to this observed PoHT hypertension are due to functional as well as structural alterations, which are partly reversible but can definitely be modulated, and druggable targets might change the cause of the disease.
We believe it’s important because hypoxia, which is the consequence of these alterations, is a strong driver of further progression to further fibrosis and Nash.
I hope you all enjoyed my talk. If there are any questions, this is a webinar. You cannot ask them directly, but don’t hesitate to reach out.
Thank you.



